Graph-based Random Walk
This notebook is used to construct a Human-Mouse graph through Random Walk method.
Introduction
Human diffusion-based tractography
We utilized diffusion data from the Human Connectome Project (HCP), which have been preprocessed following HCP’s standard pipeline.
Further post-processing steps of tractography were carried out using MRtrix3.
Mouse Tracer
Tracer data maps neural connectivity by tracking the movement of injected tracers along axonal pathways in animal brains.
The mouse connectome data we used was initially sourced from the Allen Mouse Connectivity Atlas.
Embedding space generated from Human-Mouse graph
Intra-species edges are defined by anatomical connectivity, derived from mouse viral tracer data and human tractography.
Cross-species edges were established using transcriptomic latent embeddings from DNN model, constrained by coarse-scale anatomical hierarchies.
For the pruned Human-Mouse graph, we used the Node2vec algorithm by Sporns et al. to construct graph embedding.
Define human and mouse ROIs
[1]:
%matplotlib inline
[2]:
human_select_cortex_region = ['A8m', 'A8dl', 'A9l', 'A6dl','A6m','A9m', 'A10m', 'A9/46d','IFJ', 'A46', 'A9/46v', 'A8vl','A6vl','A10l', 'A44d', 'IFS', 'A45c', 'A45r', 'A44op','A44v', 'A14m', 'A12/47o', 'A11l','A11m', 'A13', 'A12/47l','A32p','A32sg','A24cd','A24rv','A4hf', 'A6cdl', 'A4ul', 'A4t', 'A4tl', 'A6cvl','A1/2/3ll', 'A4ll','A1/2/3ulhf', 'A1/2/3tonIa', 'A2','A1/2/3tru','A7r', 'A7c', 'A5l', 'A7pc', 'A7ip', 'A39c', 'A39rd', 'A40rd', 'A40c', 'A39rv','A40rv',
'A7m', 'A5m', 'dmPOS','A31','A23d','A23c','A23v','cLinG', 'rCunG','cCunG', 'rLinG', 'vmPOS', 'mOccG', 'V5/MT+', 'OPC', 'iOccG', 'msOccG', 'lsOccG',
'G', 'vIa', 'dIa', 'vId/vIg', 'dIg', 'dId','A38m', 'A41/42', 'TE1.0 and TE1.2', 'A22c', 'A38l', 'A22r', 'A21c', 'A21r', 'A37dl', 'aSTS', 'A20iv', 'A37elv', 'A20r', 'A20il', 'A37vl', 'A20cl','A20cv', 'A20rv', 'A37mv', 'A37lv', 'A35/36r', 'A35/36c', 'lateral PPHC', 'A28/34', 'TH','TI','rpSTS','cpSTS']
mouse_select_cortex_region = ['ACAd', 'ACAv', 'PL','ILA', 'ORBl', 'ORBm', 'ORBvl','MOp','SSp-n', 'SSp-bfd', 'SSp-ll', 'SSp-m',
'SSp-ul', 'SSp-tr', 'SSp-un','SSs','PTLp','RSPagl','RSPd', 'RSPv','VISpm','VISp','VISal','VISl','VISpl','AId','AIp','AIv','GU','VISC','TEa', 'PERI', 'ECT','AUDd', 'AUDp',
'AUDpo', 'AUDv']
[3]:
len(human_select_cortex_region)
[3]:
105
[4]:
len(mouse_select_cortex_region)
[4]:
37
[5]:
human_select_subcortex_region = ['mAmyg', 'lAmyg', 'CA1', 'CA4DG', 'CA2CA3', 'subiculum','Claustrum', 'head of caudate', 'body of caudate', 'Putamen',
'posterovemtral putamen', 'nucleus accumbens','external segment of globus pallidus','internal segment of globus pallidus', 'mPMtha', 'Stha','cTtha', 'Otha',
'mPFtha','lPFtha','rTtha', 'PPtha']
mouse_select_subcortex_region = ['LA', 'BLA', 'BMA', 'PA','CA1', 'CA2', 'CA3', 'DG', 'SUB', 'ACB', 'CP', 'FS', 'SF', 'SH','sAMY', 'PAL', 'VENT', 'SPF', 'SPA', 'PP', 'GENd', 'LAT', 'ATN',
'MED', 'MTN', 'ILM', 'GENv', 'EPI', 'RT']
[6]:
len(human_select_cortex_region+human_select_subcortex_region)
[6]:
127
[7]:
Human_select_region = human_select_cortex_region + human_select_subcortex_region
Mouse_select_region = mouse_select_cortex_region + mouse_select_subcortex_region
[8]:
len(Human_select_region)
[8]:
127
[9]:
len(Mouse_select_region)
[9]:
66
[10]:
import warnings
warnings.filterwarnings('ignore')
import os
import pandas as pd
import numpy as np
import seaborn as sns
from matplotlib import pyplot as plt
import pickle
Load human structural connectivity data and region info table
[11]:
Human_dti_dataframe=pd.read_csv('../../tutorials/structural_connection/human_127atlas_dti.csv')
[12]:
Human_dti_dataframe.set_index('Unnamed: 0',drop=True,inplace=True)
[13]:
human_127atlas = pd.read_excel('../../transbrain/atlas/roi_of_bn_atlas.xlsx')
[17]:
human_127atlas
[17]:
| Anatomical Name | Full Name | Atlas Type | Left Index | Right Index | Atlas Index | |
|---|---|---|---|---|---|---|
| 0 | A8m | A8m, medial area 8 | BN | 1 | 2 | 1 |
| 1 | A8dl | A8dl, dorsolateral area 8 | BN | 3 | 4 | 2 |
| 2 | A9l | A9l, lateral area 9 | BN | 5 | 6 | 3 |
| 3 | A6dl | A6dl, dorsolateral area 6 | BN | 7 | 8 | 4 |
| 4 | A6m | A6m, medial area 6 | BN | 9 | 10 | 5 |
| ... | ... | ... | ... | ... | ... | ... |
| 122 | rTtha | rTtha, rostral temporal thalamus | BN | 237 | 238 | 123 |
| 123 | PPtha | PPtha, posterior parietal thalamus | BN | 239 | 240 | 124 |
| 124 | Otha | Otha, occipital thalamus | BN | 241 | 242 | 125 |
| 125 | cTtha | cTtha, caudal temporal thalamus | BN | 243 | 244 | 126 |
| 126 | lPFtha | lPFtha, lateral pre-frontal thalamus | BN | 245 | 246 | 127 |
127 rows × 6 columns
[14]:
Human_dti_dataframe.index = human_127atlas['Anatomical Name'].values.tolist()
Human_dti_dataframe.columns = human_127atlas['Anatomical Name'].values.tolist()
[15]:
Human_dti_dataframe = Human_dti_dataframe[Human_select_region]
Human_dti_dataframe = Human_dti_dataframe.T[Human_select_region]
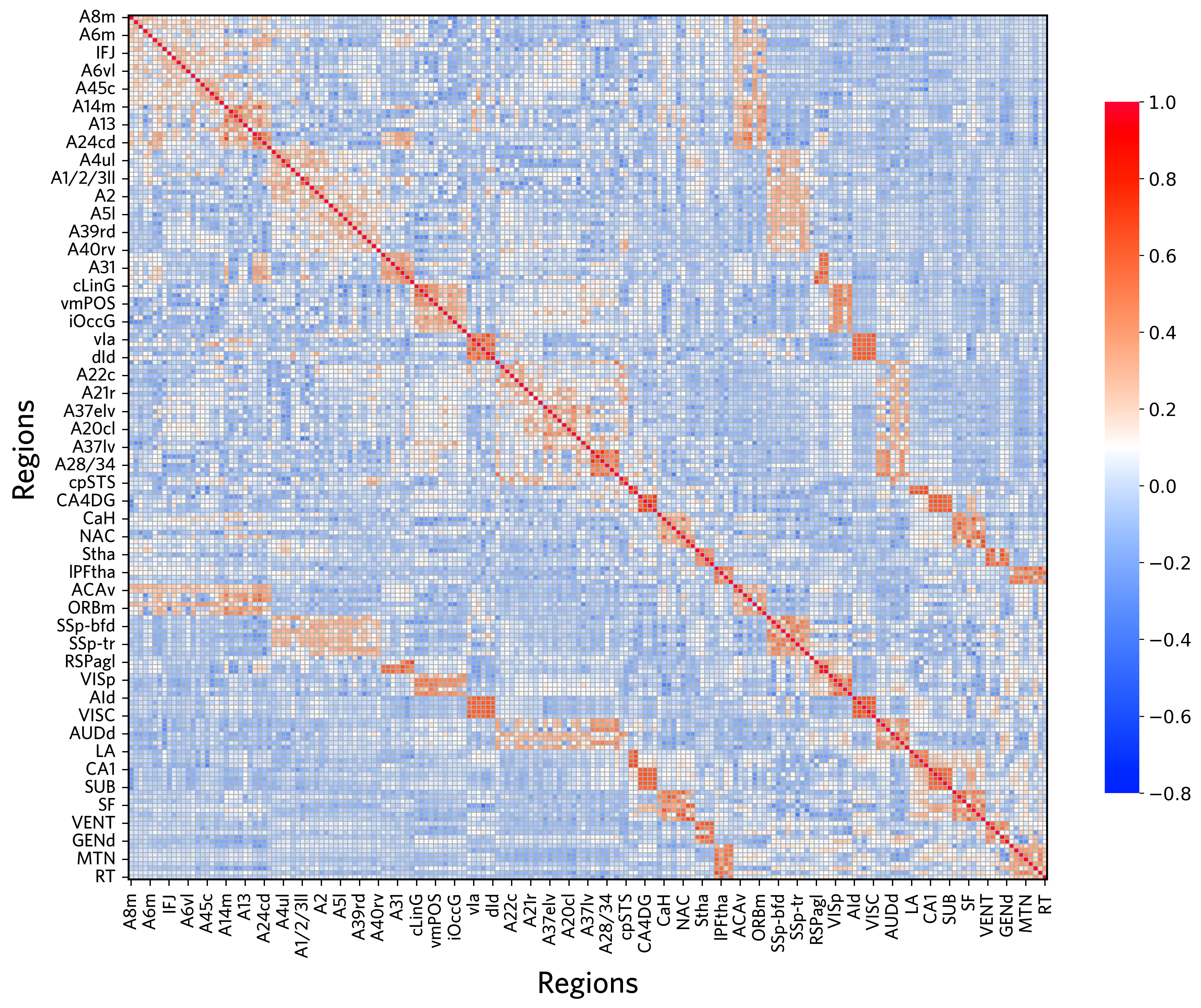
Visualize the structural connectivity matrix
[16]:
f, ax = plt.subplots(figsize=(11, 9),dpi=600)
sns.heatmap(Human_dti_dataframe,linewidths=0.3,cmap='RdBu_r',vmin=0)
ax.set(xlabel="", ylabel="")
[16]:
[Text(0.5, 421.3333333333332, ''), Text(652.3333333333331, 0.5, '')]
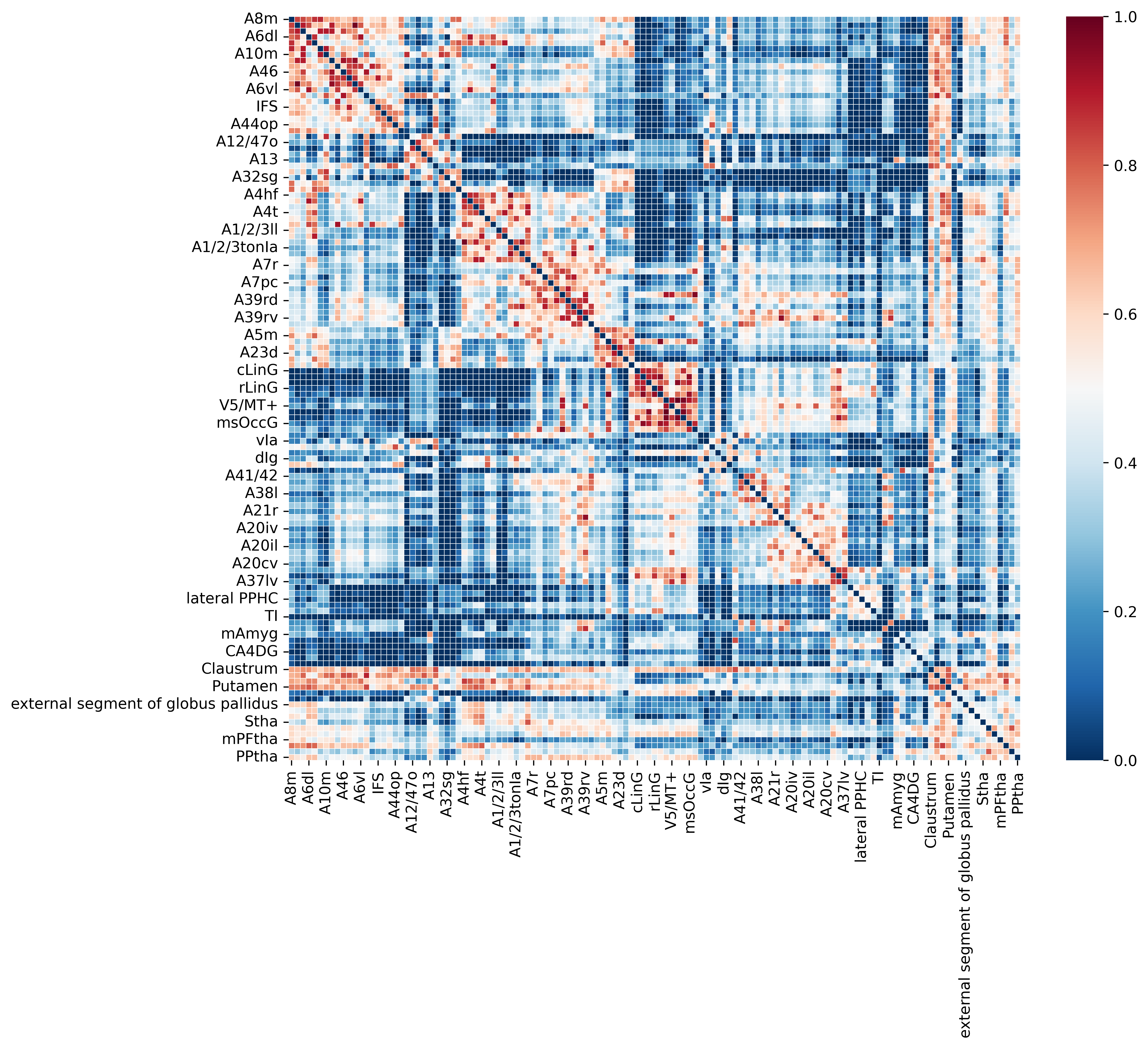
Load mouse tracer data
[19]:
Mouse_tracer_dataframe = pd.read_csv('../../tutorials/structural_connection/mouse_atlas_tracer.csv')
[20]:
Mouse_tracer_dataframe.set_index('Unnamed: 0',drop=True,inplace=True)
[21]:
Mouse_tracer_dataframe = Mouse_tracer_dataframe[Mouse_select_region]
Mouse_tracer_dataframe = Mouse_tracer_dataframe.T[Mouse_select_region]
Visualize the mouse tracer matrix
[22]:
f, ax = plt.subplots(figsize=(11, 9),dpi=600)
sns.heatmap(Mouse_tracer_dataframe,linewidths=0.3,cmap='RdBu_r',vmin=0)
[22]:
<Axes: xlabel='Unnamed: 0'>

Load anatomical hierachy constraint information
[83]:
hier_table = pd.read_excel('./files/hierarchical_brain_relationships.xlsx')
[84]:
cortex_similarity_dataframe = pd.read_csv('./files/net_emd_mean_dataframe_cortex.csv') # Region-specific TR similarity from embeddings of detached model
cortex_similarity_dataframe.set_index('Unnamed: 0',inplace=True,drop=True)
cortex_similarity_dataframe = cortex_similarity_dataframe[human_select_cortex_region]
cortex_similarity_dataframe = cortex_similarity_dataframe.T[mouse_select_cortex_region].T
[85]:
subcortex_similarity_dataframe = pd.read_csv('./files/net_emd_mean_dataframe_subcortex.csv')
subcortex_similarity_dataframe.set_index('Unnamed: 0',inplace=True,drop=True)
subcortex_similarity_dataframe = subcortex_similarity_dataframe.T[mouse_select_subcortex_region].T
[86]:
corr_matrix_mean = np.zeros((len(Human_select_region),len(Mouse_select_region)))
corr_matrix_mean[:len(human_select_cortex_region),:len(mouse_select_cortex_region)] = cortex_similarity_dataframe.T.values
corr_matrix_mean[len(human_select_cortex_region):,len(mouse_select_cortex_region):] = subcortex_similarity_dataframe.T.values
corr_matrix_mean_dataframe = pd.DataFrame(corr_matrix_mean,index=Human_select_region,columns=Mouse_select_region)
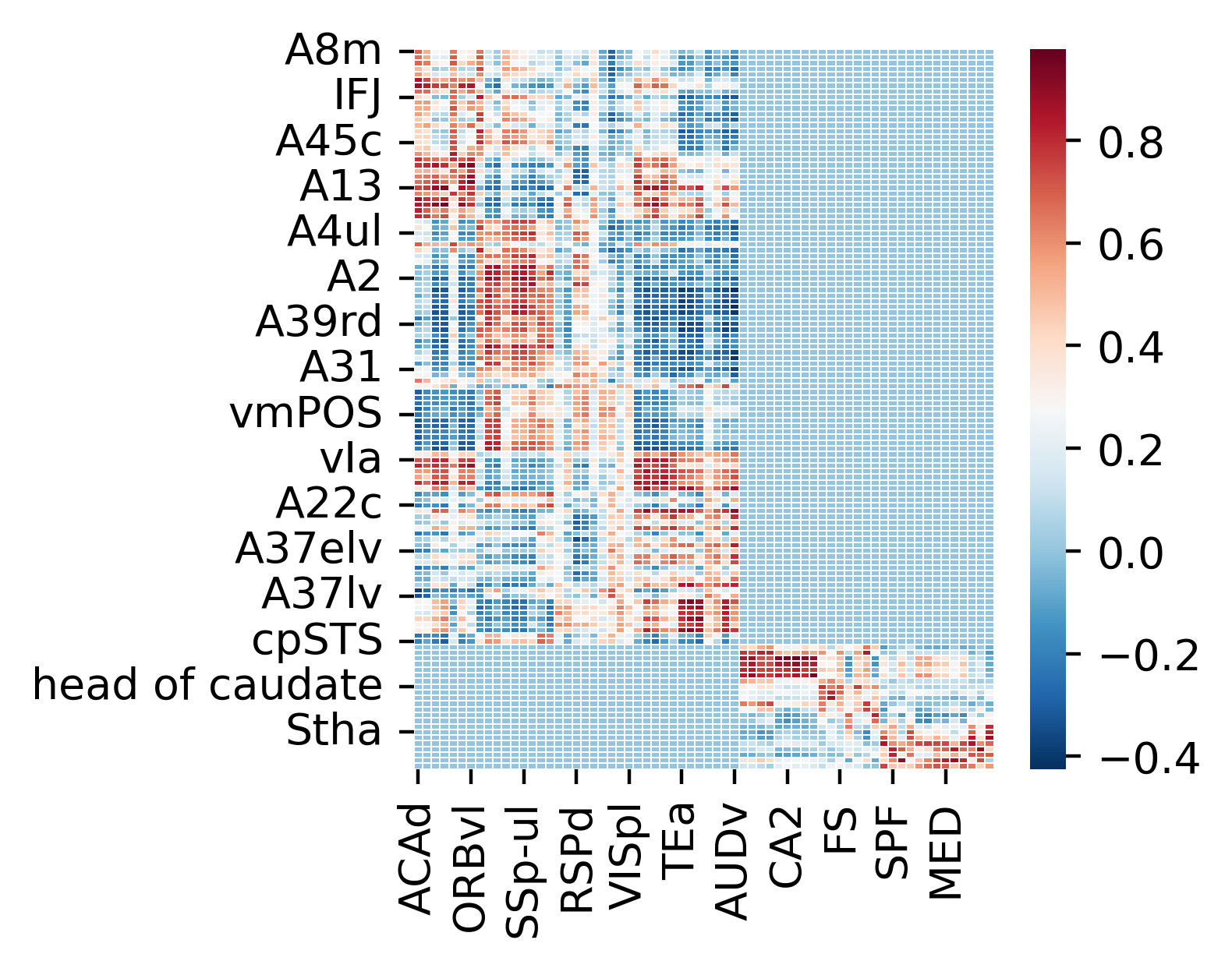
Visualization of cross-species transcriptional similarity with hierarchical constraints
[87]:
fig,ax=plt.subplots(1,1,figsize=(3,3),dpi=400)
sns.heatmap(corr_matrix_mean_dataframe,cmap='RdBu_r',linewidths=0.3)
ax.set(xlabel="", ylabel="")
[87]:
[Text(0.5, 16.888888888888864, ''), Text(34.888888888888864, 0.5, '')]
[88]:
Sparse_matrix = np.zeros_like(corr_matrix_mean_dataframe.values)
[89]:
Sparse_matrix_dataframe=pd.DataFrame(Sparse_matrix)
[90]:
Sparse_matrix_dataframe.index = corr_matrix_mean_dataframe.index
Sparse_matrix_dataframe.columns = corr_matrix_mean_dataframe.columns
[91]:
for i in range(11):
for h_n in [item.strip().strip("'") for item in hier_table['Human Regions'].iloc[i].split(",")]:
for m_n in [item.strip().strip("'") for item in hier_table['Mouse Regions'].iloc[i].split(",")]:
Sparse_matrix_dataframe.loc[h_n,m_n] = corr_matrix_mean_dataframe.loc[h_n,m_n]
[92]:
Sparse_matrix[Sparse_matrix<0]=0
[93]:
fig,ax=plt.subplots(1,1,figsize=(3,3),dpi=400)
sns.heatmap(Sparse_matrix_dataframe,cmap='RdBu_r',linewidths=0.3,vmin=0)
ax.set(xlabel="", ylabel="")
[93]:
[Text(0.5, 16.888888888888864, ''), Text(34.888888888888864, 0.5, '')]

[94]:
embedding_input=np.zeros((193,193))
embedding_input[:127,:127] = Human_dti_dataframe.values
embedding_input[:127,127:] = Sparse_matrix_dataframe.values
embedding_input[127:,:127] = Sparse_matrix_dataframe.T.values
embedding_input[127:,127:] = Mouse_tracer_dataframe.values
[95]:
region_labels = human_select_cortex_region + ['mAmyg', 'lAmyg', 'CA1', 'CA4DG', 'CA2CA3', 'subiculum', 'Claustrum','CaH','CaB', 'Putamen','PVP','NAC',
'GPe','GPi', 'mPMtha','Stha', 'cTtha', 'Otha','mPFtha', 'lPFtha', 'rTtha', 'PPtha'] + Mouse_select_region
[ ]:
'mPMtha', 'Stha','cTtha', 'Otha',
'mPFtha','lPFtha','rTtha', 'PPtha']
[96]:
embedding_input = pd.DataFrame(embedding_input,index=region_labels,columns=region_labels)
[97]:
from matplotlib import pyplot as plt
import seaborn as sns
from matplotlib.colors import LinearSegmentedColormap,Normalize
import matplotlib.colors as mcolors
import matplotlib
from matplotlib.font_manager import FontProperties
matplotlib.rcParams['axes.spines.right'] = False
matplotlib.rcParams['axes.spines.top'] = False
font_path ='../../tutorials/whitney-medium.otf'
custom_font = FontProperties(fname=font_path)
from matplotlib.patches import Rectangle
[98]:
from matplotlib import pyplot as plt
import matplotlib.colors as mcolors
from matplotlib.colors import LinearSegmentedColormap,Normalize
from matplotlib.colors import LinearSegmentedColormap, rgb_to_hsv, hsv_to_rgb
[99]:
original_cmap = plt.get_cmap("coolwarm")
colors = original_cmap(np.linspace(0, 1, 256))
def adjust_color(color, factor):
r, g, b, a = color
hsv = rgb_to_hsv([r, g, b])
hsv[1] = min(1, hsv[1] * factor)
hsv[2] = min(1, hsv[2] * factor)
adjusted_rgb = hsv_to_rgb(hsv)
return np.array([adjusted_rgb[0], adjusted_rgb[1], adjusted_rgb[2], a])
red_adjustment = 1.5
blue_adjustment = 1.5
for i in range(len(colors)):
if colors[i, 2] > 0.5:
colors[i] = adjust_color(colors[i], blue_adjustment)
elif colors[i, 0] > 0.5:
colors[i] = adjust_color(colors[i], red_adjustment)
custom_cmap = LinearSegmentedColormap.from_list("custom_coolwarm_adjusted", colors)
[100]:
def brighten_cmap(cmap, factor=1.2):
new_colors = []
for c in cmap(np.linspace(0, 1, cmap.N)):
new_color = mcolors.rgb_to_hsv(c[:3])
new_color[2] = min(new_color[2] * factor, 1.0)
new_colors.append(mcolors.hsv_to_rgb(new_color))
return LinearSegmentedColormap.from_list("bright_plasma", new_colors)
bright_cmap = brighten_cmap(custom_cmap, factor=6)
Visualization of the initial cross-species graph
[101]:
f, ax = plt.subplots(figsize=(14, 11), dpi=300)
heatmap = sns.heatmap(
embedding_input,
cmap=bright_cmap,
vmin=-0.2,
vmax=1,
linewidths=0.3,
linecolor='gray',
cbar_kws={
"orientation": "vertical",
"shrink": 0.8,
"aspect": 20,
"label": "",
"ticks": [0, 0.5, 1.0]
}
)
cbar = heatmap.collections[0].colorbar
cbar.ax.tick_params(labelsize=14)
ax.set_xlabel("Regions", fontsize=24, labelpad=10, fontproperties=custom_font)
ax.set_ylabel("Regions", fontsize=24, labelpad=10, fontproperties=custom_font)
ax.set_xticklabels(ax.get_xticklabels(), fontsize=10, rotation=90, fontproperties=custom_font)
ax.set_yticklabels(ax.get_yticklabels(), fontsize=10, rotation=0, fontproperties=custom_font)
ax.tick_params(axis='both', which='both', length=5, width=1, pad=5)
plt.xticks(fontsize=14)
plt.yticks(fontsize=14)
for spine in ax.spines.values():
spine.set_visible(True)
spine.set_color('black')
spine.set_linewidth(1.5)
plt.tight_layout()
plt.show()
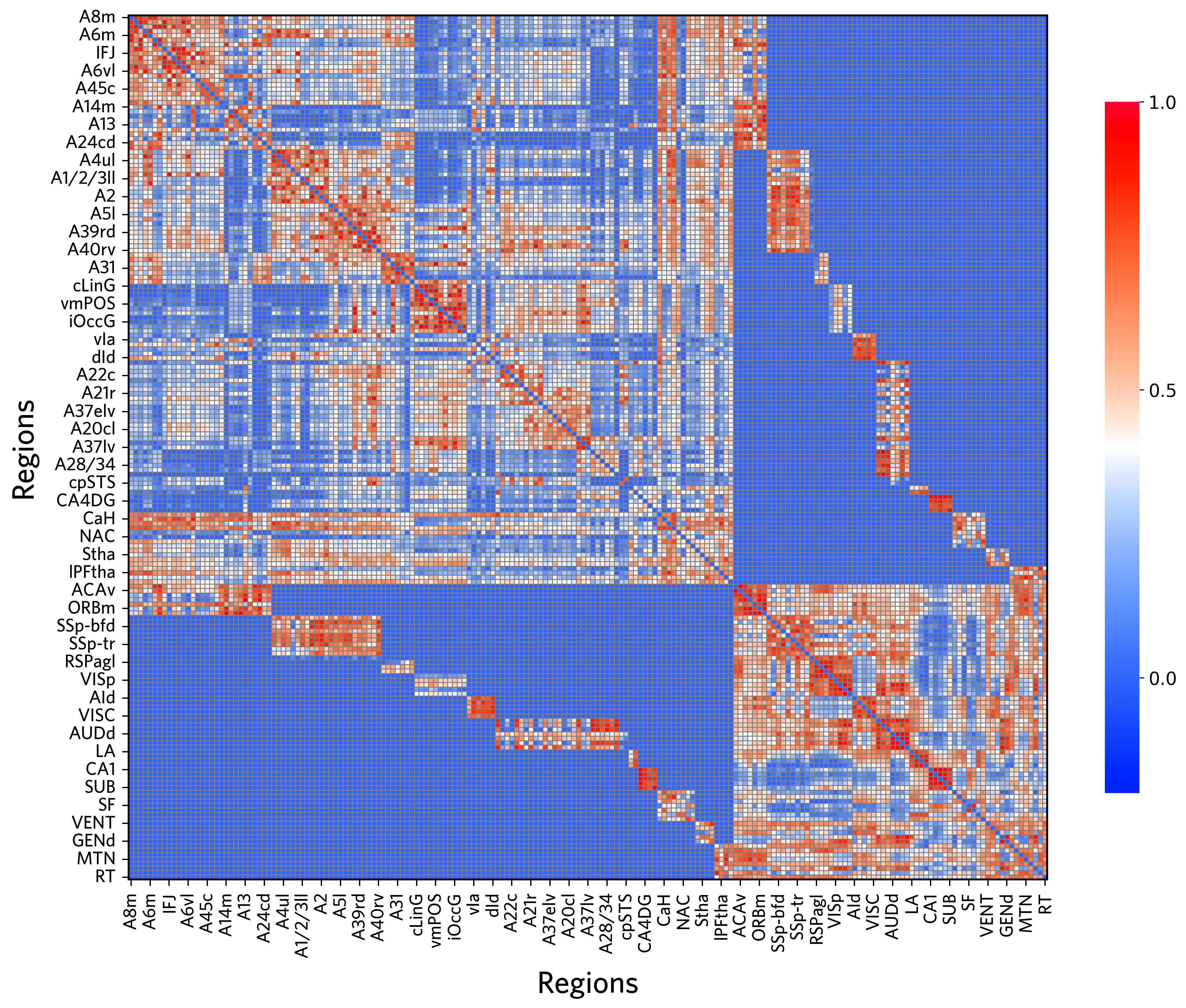
[102]:
embedding_input_dataframe=pd.DataFrame(embedding_input)
# embedding_input_dataframe.to_csv('mouse_human_embedding_input.csv')
[1]:
#! python ../code/graph_walk/run.py -i ./mouse_human_embedding_input.csv -s graph_embedding -p 0.01 -q 0.1 -pm 500 # Generate graph embeddings
[104]:
def get_embedding_map(p,q):
with open("./graph_embedding/Human_Mouse_p{}_q{}_graph_embeddings.pkl".format(p,q),'rb') as f:
embedding_data=pickle.load(f)
embedding_zero=np.zeros((500,386,386))
for i in range(500):
embeeding_corr=np.corrcoef(embedding_data[i],embedding_data[i])
embedding_zero[i]=embeeding_corr
aver_embedding=np.mean(embedding_zero,axis=0)
embedding_map=pd.DataFrame(aver_embedding[:193,193:])
return embedding_map
[105]:
embedding_map=get_embedding_map(0.01,0.1)
[106]:
embedding_map.index = region_labels
embedding_map.columns = region_labels
Visualization of final obtained cross-species average graph embeddings
[107]:
f, ax = plt.subplots(figsize=(14, 11), dpi=300)
heatmap = sns.heatmap(
embedding_map,
cmap= bright_cmap,
vmin=-0.8,
vmax=1,
linewidths=0.3,
linecolor='#B0B0B0',
cbar_kws={
"orientation": "vertical",
"shrink": 0.8,
"aspect": 20,
"label": ""
}
)
cbar = heatmap.collections[0].colorbar
cbar.ax.tick_params(labelsize=14)
ax.set_xlabel("Regions", fontsize=24, labelpad=10, fontproperties=custom_font)
ax.set_ylabel("Regions", fontsize=24, labelpad=10, fontproperties=custom_font)
ax.set_xticklabels(ax.get_xticklabels(), fontsize=10, rotation=90, fontproperties=custom_font)
ax.set_yticklabels(ax.get_yticklabels(), fontsize=10, rotation=0, fontproperties=custom_font)
ax.tick_params(axis='both', which='both', length=5, width=1, pad=5)
plt.xticks(fontsize=14)
plt.yticks(fontsize=14)
for spine in ax.spines.values():
spine.set_visible(True)
spine.set_color('black')
spine.set_linewidth(1.5)
plt.tight_layout()
plt.show()