Basic use 2: Human to mouse
Here is a brief demonstration on how to use TransBrain to map the phenptype from human to mouse. The example data used in this tutorial can be download from exampledata directory of our GitHub repository.
Step 1: Prepare input data
There are two ways to input data. One is that you already have region-level phenotype data as a CSV table which follows the format and region order in the provided template file. The DataFrame contains two columns: ['Anatomical Name', 'Phenotype'].
Here we load the example human data table.
[53]:
import pandas as pd
# Example human data
human_phenotype = pd.read_csv('../../../transbrain/exampledata/human/bn/human_bn_all_example_data.csv',index_col=0)
[54]:
human_phenotype
[54]:
| Phenotype | |
|---|---|
| Anatomical Name | |
| A8m | 0.260696 |
| A8dl | -0.132093 |
| A9l | -0.841146 |
| A6dl | 0.151087 |
| A6m | 0.614044 |
| ... | ... |
| Otha | 2.237752 |
| mPFtha | 2.532931 |
| lPFtha | 2.300108 |
| rTtha | 2.957160 |
| PPtha | 1.973803 |
127 rows × 1 columns
Or if you data is volumetric data in .nii or .nii.gz format that has been aligned to the human atlas space. You can use the following functions to extract region-level data. When fetching atlas, you need to choose the region_type parameter from {‘cortex’, ‘subcortex’, ‘all’}, which controls the regions to include in returned region names and info table.
[55]:
import transbrain as tb
help(tb.atlas.fetch_human_atlas)
Help on function fetch_human_atlas in module transbrain.atlas:
fetch_human_atlas(atlas_type: Literal['bn', 'dk', 'aal'] = 'bn', region_type: Literal['cortex', 'subcortex', 'all'] = 'all')
Fetch a human brain atlas image and its region information.
This function loads a labeled brain atlas image (e.g., Brainnetome, Desikan-Killiany, or AAL)
along with its corresponding ROI (region of interest) metadata table.
Parameters
----------
atlas_type : {'bn', 'dk', 'aal'}, optional
The type of atlas to load. Must be one of:
- 'bn' : Brainnetome Atlas
- 'dk' : Desikan-Killiany Atlas
- 'aal' : Automated Anatomical Labeling (AAL) Atlas
Default is 'bn'.
region_type : {'cortex', 'subcortex', 'all'}, optional
Which regions to include in returned region names and info table: cortical, subcortical, or all. Default is 'all'.
Returns
-------
dict
A dictionary with the following keys:
- 'atlas' : nibabel.Nifti1Image
The loaded NIfTI image of the atlas.
- 'atlas_data' : np.ndarray
The atlas volume data as a NumPy array.
- 'region_info' : np.ndarray
A list of anatomical region names, extracted from the 'Anatomical Name' column of the label file.
- 'info_table' : pandas.DataFrame
The full region-of-interest (ROI) information table including additional metadata.
Raises
------
FileNotFoundError
If the atlas image file or the ROI label file is not found.
[56]:
#Fetch human atlas
human_atlas = tb.atlas.fetch_human_atlas(atlas_type='bn',region_type='all')
[57]:
human_atlas.keys()
[57]:
dict_keys(['atlas', 'atlas_data', 'region_info', 'info_table'])
The first two items in the returned dictionary are the NIfTI file of the atlas and the array in it, while the next two items store the brain region names and a table containing label values.
[58]:
human_atlas['info_table']
[58]:
| Anatomical Name | Atlas Index | |
|---|---|---|
| 0 | A8m | 1 |
| 1 | A8dl | 2 |
| 2 | A9l | 3 |
| 3 | A6dl | 4 |
| 4 | A6m | 5 |
| ... | ... | ... |
| 122 | rTtha | 123 |
| 123 | PPtha | 124 |
| 124 | Otha | 125 |
| 125 | cTtha | 126 |
| 126 | lPFtha | 127 |
127 rows × 2 columns
Next, we use the get_region_phenotypes function to calculate regional phenotype data from the image. In addition to the phenotype image and the atlas dictionary, you also need to specify the atlas name and the regions to be extracted.
[59]:
help(tb.base.get_region_phenotypes)
Help on function get_region_phenotypes in module transbrain.base:
get_region_phenotypes(phenotype_nii_path: str, atlas_dict: dict, atlas_type: Literal['bn', 'dk', 'aal', 'mouse'] = 'bn', region_type: Literal['cortex', 'subcortex', 'all'] = 'all', method: str = 'mean', resample: bool = True, label_column: str = 'Atlas Index', region_column: str = 'Anatomical Name') -> pandas.core.frame.DataFrame
Calculate region-wise phenotype values using a specified brain atlas.
This function extracts regional statistics (mean or sum) from a phenotype NIfTI image
based on a chosen human or mouse brain atlas. The atlas can be automatically
resampled to match the phenotype image resolution if needed.
Parameters
----------
phenotype_nii_path : str
Path to the input phenotype NIfTI file. Should be in MNI space for human atlases,
or Allen CCFv3 space for mouse atlas.
atlas_dict : dict
A dictionary containing the following keys:
- 'atlas': The loaded Mouse atlas image.
- 'atlas_data': The atlas data as a numpy array.
- 'region_info': A list of anatomical names for the specified regions.
- 'info_table': The full ROI information table.
atlas_type : {'bn', 'dk', 'aal','mouse}, optional
The type of atlas. Must be one of:
- 'bn' : Brainnetome Atlas (default)
- 'dk' : Desikan-Killiany Atlas
- 'aal' : Automated Anatomical Labeling (AAL) Atlas
- 'mouse' : Allen Mouse CCFv3 atlas
region_type : {'cortex', 'subcortex', 'all'}, optional
Which regions to include in returned region names and info table: cortical, subcortical, or all. Default is 'all'.
method : {'mean', 'sum'}, optional
Method for aggregating voxel values within each region. Default is 'mean'.
resample : bool, optional
If True, resample the atlas to match the shape and resolution of the phenotype image.
Default is True.
label_column : str, optional
Name of the column in the atlas label CSV that contains numeric label indices.
Default is 'Atlas Index'.
region_column : str, optional
Name of the column in the atlas label CSV that contains region names.
Default is 'Anatomical Name'.
Returns
-------
pandas.DataFrame
A DataFrame with aggregated phenotype values (mean or sum) for each region, , indexed by brain region name.
[60]:
# Get phenotypes in Human atlas used in TransBrain
phenotype_nii_path = '../../../transbrain/exampledata/human/human_example_phenotype_data.nii.gz'
human_phenotype = tb.base.get_region_phenotypes(phenotype_nii_path, atlas_dict = human_atlas, atlas_type='bn', region_type='all')
[61]:
human_phenotype
[61]:
| Phenotype | |
|---|---|
| Anatomical Name | |
| A8m | 0.260747 |
| A8dl | -0.132129 |
| A9l | -0.841126 |
| A6dl | 0.151081 |
| A6m | 0.614070 |
| ... | ... |
| Otha | 2.237802 |
| mPFtha | 2.532970 |
| lPFtha | 2.300147 |
| rTtha | 2.957121 |
| PPtha | 1.973806 |
127 rows × 1 columns
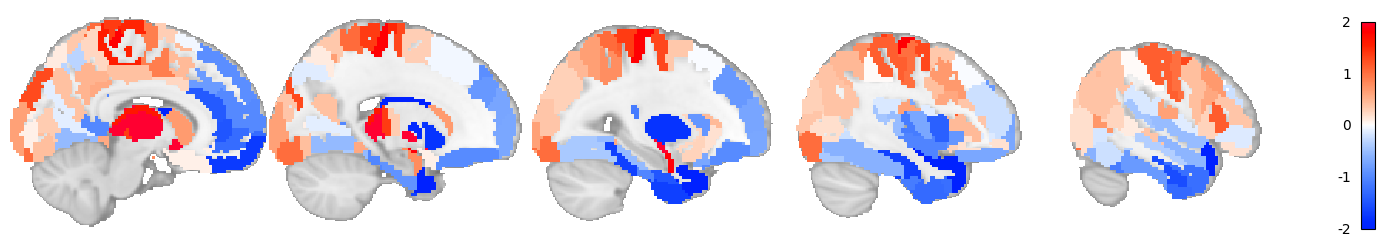
Visualizing human phenotype data
Using visualization function of TransBrain to view the data.
[62]:
from transbrain.vis import map_phenotype_to_nifti, plot_human_phenotype
# map the region-level phenotype data to an image
phenotype_img = map_phenotype_to_nifti(human_phenotype, human_atlas)
[63]:
# view the image
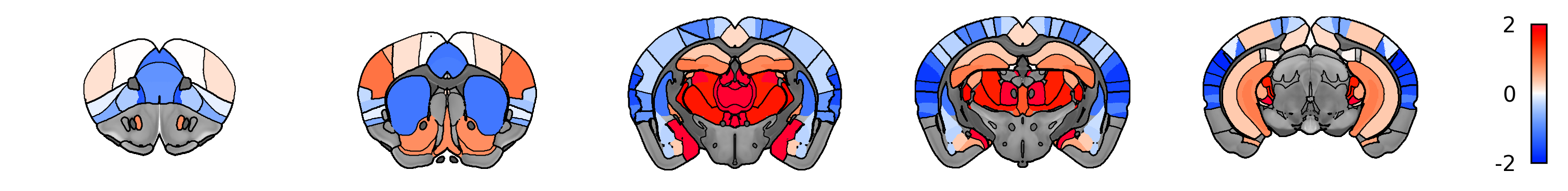
plot_human_phenotype(phenotype_img, normalize_img=True, cut_coords=range(10, 60, 10), vmax=2, symmetric_cbar = True)
<Figure size 3000x300 with 0 Axes>
Step 2: Initialization
Before starting the mapping, you need to initialize TransBrain by creating an instance of the SpeciesTrans class.
You need to specify which human brain atlas space you want to map from to the mouse brain. You can choose atlas_type from ['bn', 'dk', 'aal']. During this process, TransBrain will automatically load the necessary information for the mapping, including the brain region name list and the graph embedding matrix for the selected atlas.
[64]:
#Initialize TransBrain for bn atlas
Transformer = tb.trans.SpeciesTrans('bn')
INFO:root:Initialized for bn atlas.
Step 3: Mapping
Now, we can map the human phenotype DataFrame (either loaded or extracted) to mouse brain, you need to choose the regions to map and whether to normalize the data. This step is enabled by default. If your data has already been normalized, you can set it to False.
[65]:
help(Transformer.human_to_mouse)
Help on method human_to_mouse in module transbrain.trans:
human_to_mouse(phenotype: pandas.core.frame.DataFrame, region_type: Literal['cortex', 'subcortex', 'all'] = 'cortex', normalize: bool = True, restore: bool = False) -> pandas.core.frame.DataFrame method of transbrain.trans.SpeciesTrans instance
Translate human phenotype to mouse.
Parameters
----------
phenotype : pd.DataFrame
Human phenotype DataFrame (regions × phenotypes).
region_type : {'cortex', 'subcortex', 'all'}, optional
The brain region type to translate. Default is 'cortex'.
normalize : bool, optional
Whether to normalize data before translation. Default is True.
restore : bool, optional
Whether to restore values back to original scale after translation. Only used if normalize is True.
Please enable this parameter with caution, unless you are certain that the distributions of this phenotype are consistent between the two species.
Returns
-------
pd.DataFrame
Translated mouse phenotype DataFrame (regions × phenotypes).
[66]:
# Example from human to mouse
human_phenotype_in_mouse = Transformer.human_to_mouse(
human_phenotype,
region_type='all',
normalize=True
)
INFO:root:Successfully translated human all phenotypes to mouse.
Once the mapping is complete, you will see a success message. Let’s take a look at the mapped data. Now, you got the DataFrame that stores the phenotype data corresponding to the mouse CCFv3 atlas.
[67]:
human_phenotype_in_mouse
[67]:
| Phenotype | |
|---|---|
| ACAd | 0.296682 |
| ACAv | 0.303369 |
| PL | 0.327434 |
| ILA | 0.332395 |
| ORBl | 0.386486 |
| ... | ... |
| MTN | 0.696940 |
| ILM | 0.688615 |
| GENv | 0.638572 |
| EPI | 0.731165 |
| RT | 0.640975 |
68 rows × 1 columns
You can also visualize what the mapped data looks like on mouse brain.
[68]:
from transbrain.vis import plot_mouse_phenotype
#Fetch mouse atlas
mouse_atlas = tb.atlas.fetch_mouse_atlas(region_type='all')
# map the region-level phenotype data to an image
mapped_phenotype_img = map_phenotype_to_nifti(human_phenotype_in_mouse, mouse_atlas)
[69]:
# view the image
plot_mouse_phenotype(mapped_phenotype_img, normalize_img=True, symmetric_cbar=True,vmax=2, threshold=0)